Our research in
Chemical biology &
medicinal chemistry
FROM PROTEIN SURFACE RECOGNITION TO PEPTIDE THERAPEUTICS
Peptides are useful molecules to interfere with biomolecular interactions, investigate biological processes and develop new modalities for therapeutic applications. In the group, we are combining peptidomimetic and foldamer chemistries, structure-guided design and chemical biology to develop effective ligands against targets of various therapeutic interest (receptor ligands, disruptors of protein-protein interactions). In particular, we are interested in approaches to constrain peptides as they may yield compounds with unusual activity profiles including improved binding, higher resistance to degradation by proteases, and cell permeability compared with unconstrained peptides.
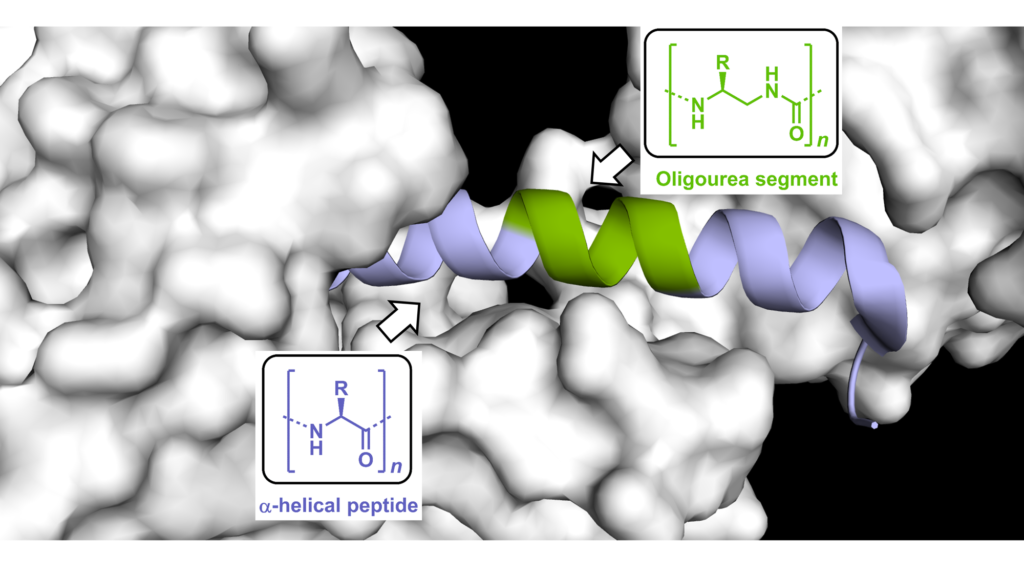
Figure : Effective mimicry of GLP-1 using a foldamer-based approach that consists in replacing a α-helical peptide portion by a oligourea insert (green).
Foldamers to target protein surfaces and improve bioactive peptides
Our ability to interface foldamers with α-eptides opens enticing opportunities for mimicking biologically active peptides and addressing some of their current limitations including stability in biological fluids (Pasco et al. in Comprehensive Supramolecular Chemistry II, 2017). However, only few unnatural helical backbone have been shown to effectively mimic biologically active peptides (e.g. for targeting protein-protein interactions or activating receptors). In 2019, we demonstrated (Nat Commun 2019, Chem Sci 2019) that peptide-oligourea helices can produce mimics of Class-B GPCR ligands with increased resistance to proteolytic degradation and prolonged duration of action in vivo. Towards the goal of developing new modalities targeted to specific proteins, we propose an integrated approach featuring combination of peptidomimetic and foldamer chemistries, structure-guided design and chemical biology to enhance the properties of biological peptides including cell uptake, and develop new inhibitors as well as molecular probes to study the consequences of target engagement in cells. One specific objective in this project is to gain details at atomic resolution of the interactions between chimeric helices and the surface of target proteins and identify general principles to guide the design of peptide/oligourea-based ligands.
Funded by :



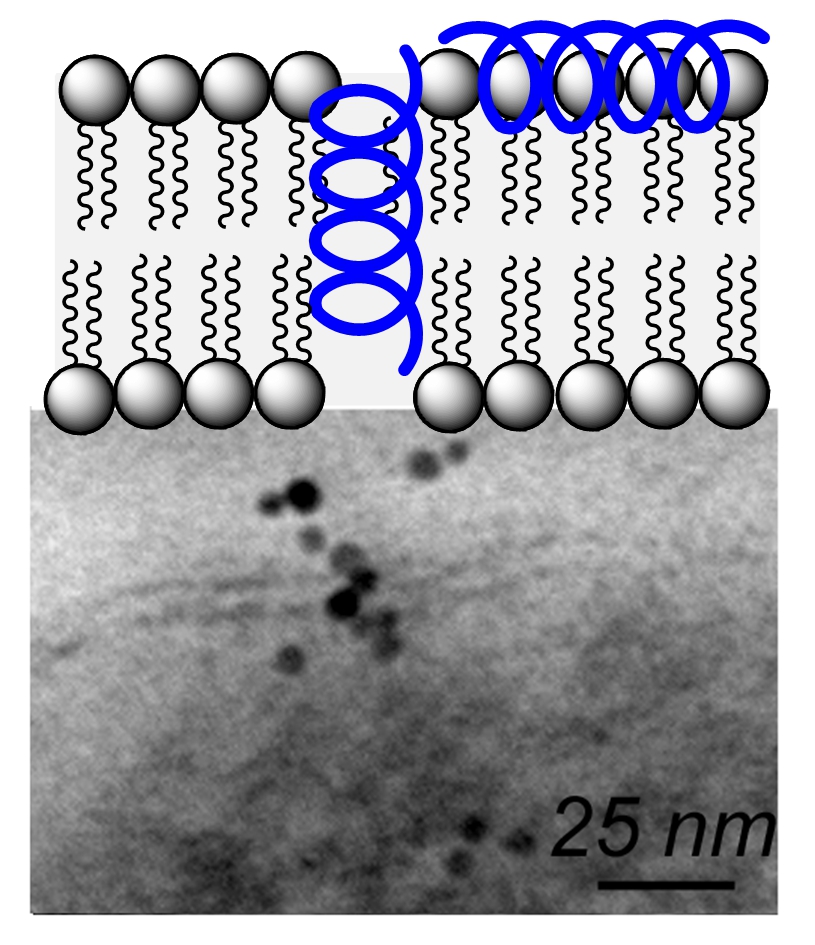
From Host defense peptide mimics to Cell Penetrating Foldamers (CPFs)
Short chain oligoureas designed to mimic globally amphiphilic cationic α-helical host-defense peptides are active against a range of Gram-negative and Gram-positive bacteria (ACIE 2010). These oligoureas which display a high helical content in the vicinity of phospholipid membranes are thought to be active by a mechanism involving permeabilization of the bacterial membrane. Such foldamers were found to be active against bacterial forms of Bacillus anthracis encountered in vivo and to exert partial protection in cutaneous and inhalational models of infection with B. anthracis (J Med Chem 2016). We found that sequence variations including the introduction of pH-responsive side chains (His type side chains) and dimerization could be used to convert these antimicrobial foldamers into gene transfection agents. Bioreducible helical cell penetrating foldamers (CPFs) with high capacity to assemble with plasmid DNA and to deliver nucleic acids into the cell have been reported (ACIE 2015). New CPF sequences with activity in the presence of serum as well as sequences for siRNA delivery were also identified (CC 2021).
Funded by :

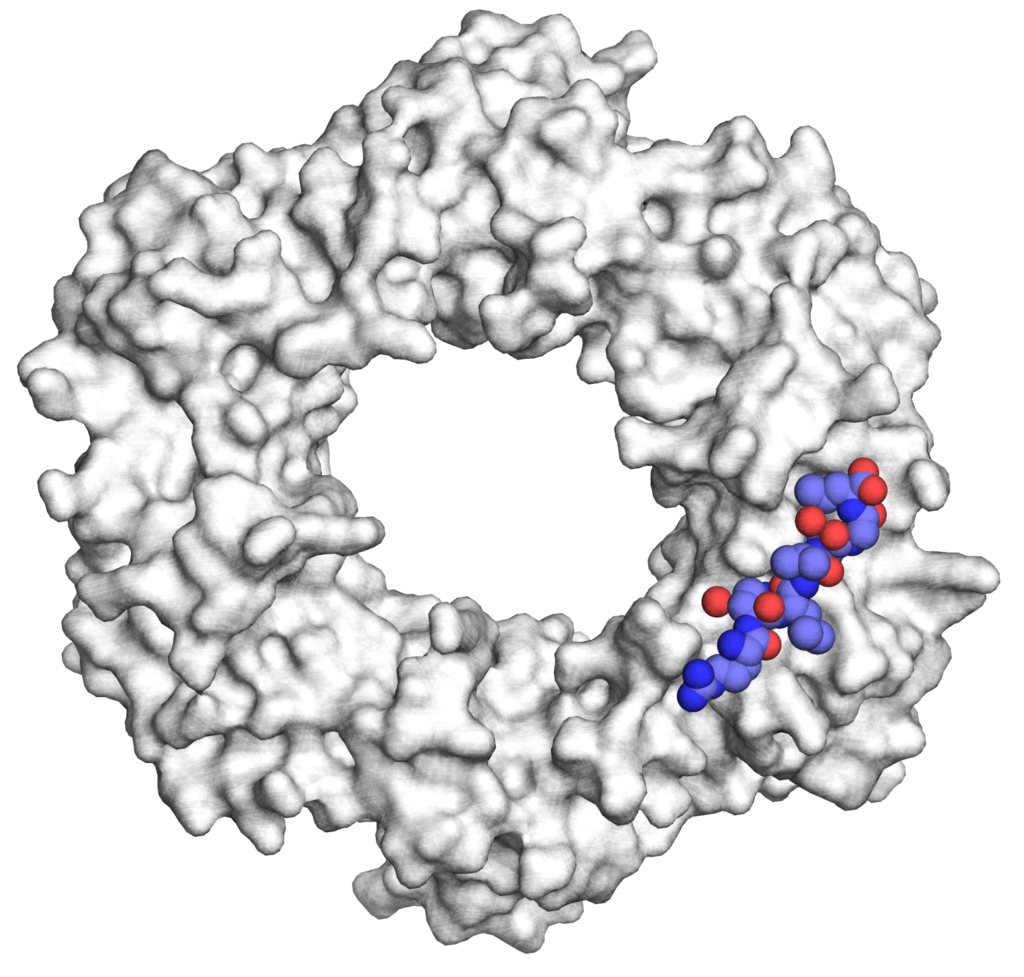
Antibacterial peptides targeting bacterial machinery : protein synthesis and DNA replication
There is an urgent need to combat drug-resistant infections and to develop potent pharmaceutical agents that are effective against multi-drug resistant pathogens. Our work focuses on the design of peptides and peptidomimetics that specifically target essential bacterial machineries, namely, DNA replication and protein synthesis (J Med Chem 2014, NSMB 2015). Because of its central function in the bacterial replisome, the replication processivity factor, or DNA sliding clamp (SC) has recently emerged as a promising target for the development of novel antibiotics. By using structure-guided design, we have designed high affinity SC ligands and have shown that they can be delivered into Gram-negative bacteria following conjugation to proline-rich antimicrobial peptides (PrAMPs) a class of antimicrobial which also target the bacterial ribosome (RSC Chem Biol, 2020).
Funded by :
